Aligning Pairs of Sequences
This example shows how to extract some sequences from GenBank®, find open reading frames (ORFs), and then align the sequences using global and local alignment algorithms.
Accessing NCBI Data from the MATLAB® Workspace
One of the many fascinating sections of the NCBI web site is the Genes and diseases section. This section provides a comprehensive introduction to medical genetics.
In this example you will be looking at genes associated with Tay-Sachs Disease. Tay-Sachs is an autosomal recessive disease caused by mutations in both alleles of a gene (HEXA, which codes for the alpha subunit of hexosaminidase A) on chromosome 15.
The NCBI reference sequence for HEXA has accession number NM_000520. You can use the getgenbank function to retrieve the sequence information from the NCBI data repository and load it into MATLAB®.
humanHEXA = getgenbank('NM_000520');
By doing a BLAST search or by searching in the mouse genome you can find an orthogonal gene, AK080777 is the accession number for a mouse hexosaminidase A gene.
mouseHEXA = getgenbank('AK080777');
For your convenience, previously downloaded sequences are included in a MAT-file. Note that data in public repositories is frequently curated and updated; therefore the results of this example might be slightly different when you use up-to-date datasets.
load('hexosaminidase.mat','humanHEXA','mouseHEXA')
Exploring the Open Reading Frames (ORFs)
You can use the function seqshoworfs to look for ORFs in the sequence for the human HEXA gene. Notice that the longest ORF is on the first reading frame. The output value in the variable humanORFs is a structure giving the position of the start and stop codons for all the ORFs on each reading frame.
humanORFs = seqshoworfs(humanHEXA.Sequence)
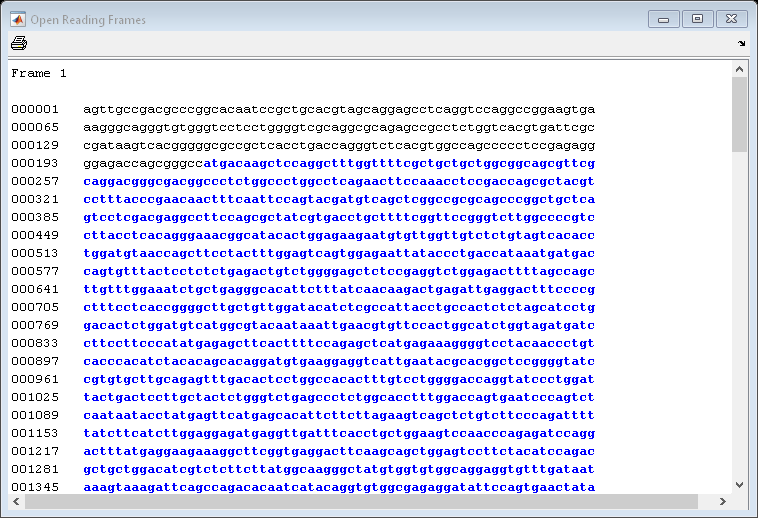
humanORFs=1×3 struct array with fields:
Start
Stop
Now look at the ORFs in the mouse HEXA gene. In this case the ORF is also on the first frame.
mouseORFs = seqshoworfs(mouseHEXA.Sequence)
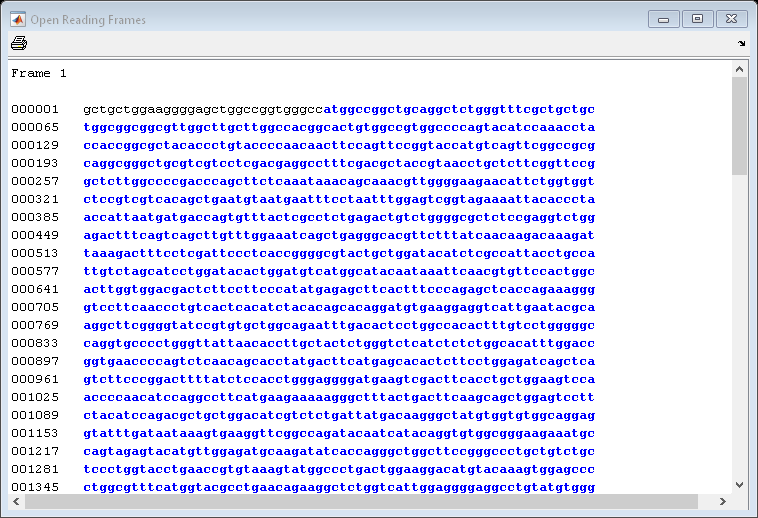
mouseORFs=1×3 struct array with fields:
Start
Stop
Aligning the Sequences
The first step is to use global sequence alignment to look for similarities between these sequences. You could look at the alignment between the nucleotide sequences, but it is generally more instructive to look at the alignment between the protein sequences, in this example we know that the sequences are coding sequences. Use the nt2aa function to convert the nucleotide sequences into the corresponding amino acid sequences. Observe that the HEXA gene occurs in the first frame for both sequences, otherwise you should use the input argument Frame to specify an alternative coding frame.
humanProtein = nt2aa(humanHEXA.Sequence); mouseProtein = nt2aa(mouseHEXA.Sequence);
One of the easiest ways to look for similarity between sequences is with a dot plot.
seqdotplot(mouseProtein,humanProtein)
Warning: Match matrix has more points than available screen pixels.
Scaling image by factors of 1 in X and 2 in Y.
xlabel('Human hexosaminidase A');ylabel('Mouse hexosaminidase A');
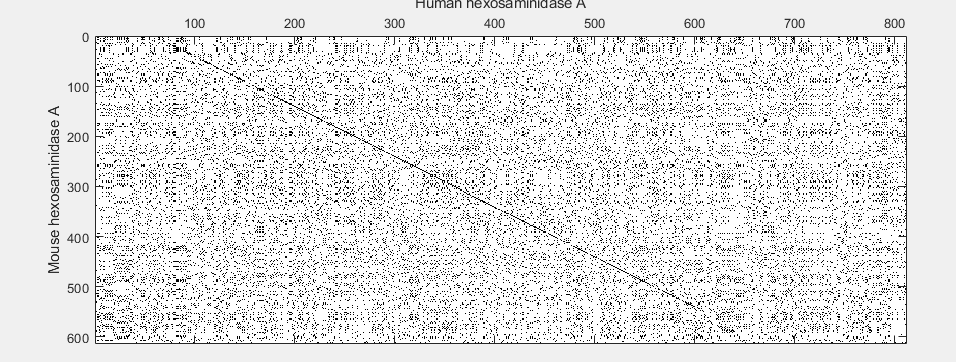
With the default settings, the dot plot is a little difficult to interpret, so you can try a slightly more stringent dot plot.
seqdotplot(mouseProtein,humanProtein,4,3)
Warning: Match matrix has more points than available screen pixels.
Scaling image by factors of 1 in X and 2 in Y.
xlabel('Human hexosaminidase A');ylabel('Mouse hexosaminidase A');
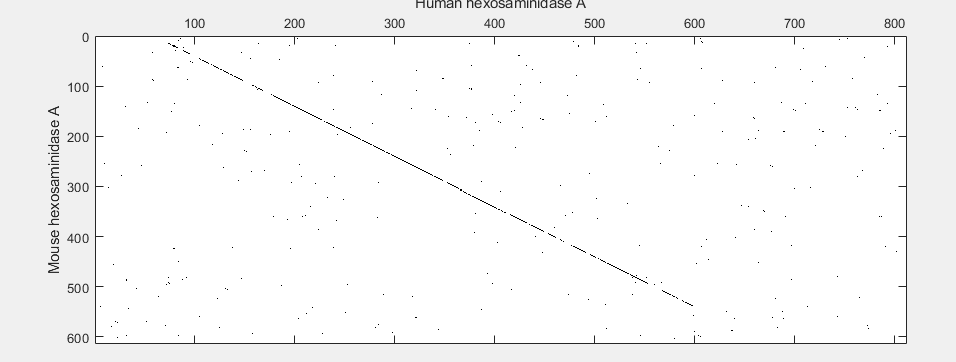
The diagonal line indicates that there is probably a good alignment so you can now take a look at the global alignment using the function nwalign which uses the Needleman-Wunsch algorithm.
[score, globalAlignment] = nwalign(humanProtein,mouseProtein)
score = 634.3333
globalAlignment = 3x812 char array
'SCRRPAQSAARSRSLRSRPEVKGQGVGPPGVAGAEPPLVT*FADKSRGRRSPDQGLTWPAPSERGDQRAMTSSRLWFSLLLAAAFAGRATALWPWPQNFQTSDQRYVLYPNNFQFQYDVSSAAQPGCSVLDEAFQRYRDLLFGSGSWPRPYLTGKRHTLEKNVLVVSVVTPGCNQLPTLESVENYTLTINDDQCLLLSETVWGALRGLETFSQLVWKSAEGTFFINKTEIEDFPRFPHRGLLLDTSRHYLPLSSILDTLDVMAYNKLNVFHWHLVDDPSFPYESFTFPELMRKGSYNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGIPGLLTPCYSGSEPSGTFGPVNPSLNNTYEFMSTFFLEVSSVFPDFYLHLGGDEVDFTCWKSNPEIQDFMRKKGFGEDFKQLESFYIQTLLDIVSSYGKGYVVWQEVFDNKVKIQPDTIIQVWREDIPVNYMKELELVTKAGFRALLSAPWYLNRISYGPDWKDFYIVEPLAFEGTPEQKALVIGGEACMWGEYVDNTNLVPRLWPRAGAVAERLWSNKLTSDLTFAYERLSHFRCELLRRGVQAQPLNVGFCEQEFEQT*APGTEEGAGCR*MVVEPGFHCILARGRSPLPSCPLPACPCAWRERGRCWRSHSIKSNVAFFYNKHGLPVFKKKSVNGVRVRAQPGWSQCLPLRSFKLRAGNETYSLCAVLPCL*AMSLPSHS*PYSRHLP*SSACSLHFCIISPRRWYMEKDVGAWRCSGQWGGLQTQPGHRRASPPCILIHLPPLELFSFGFLAASILYNHYLNIIKHILFS'
' || |: | | | | | ||:: ||| |||||||:| ||||||||| :|| :||:||||||||:| |||||| || ||||||:|||:||||||||||| :::|::|| ||:||||||| ||::|:|||||||||||||||||| |||||||||||||||||||||||||||||||:|:|||||||||:|||||||||||||||||||||||||:|||||||||| |||||||||||| ||||:|||||||||||||||||||||||||||||||||||||||||| |||||||||||: ||||||||||||:||:||||:|||:|||||||||||||||||||||||||:|| ||:|||| ||||||||||||||||||:| |||||||||||||||::||||||||||::||:|| |:: :|:|||||||||||||||::|||||||:| ||||||:||||||||||||||||||||||:||||||||||||||||||||::||::: ||::|||||||||:|||:||||::|| |||||||||| | :| : :|| | | || |: :: | | :: | : :| : | :| : : | ::: | | |::| : | | | :| ||::|| | |: | | | :: |: | '
'--------AA------------GR--------G---------A----G-R-------W----------AMAGCRLWVSLLLAAALACLATALWPWPQYIQTYHRRYTLYPNNFQFRYHVSSAAQAGCVVLDEAFRRYRNLLFGSGSWPRPSFSNKQQTLGKNILVVSVVTAECNEFPNLESVENYTLTINDDQCLLASETVWGALRGLETFSQLVWKSAEGTFFINKTKIKDFPRFPHRGVLLDTSRHYLPLSSILDTLDVMAYNKFNVFHWHLVDDSSFPYESFTFPELTRKGSFNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGAPGLLTPCYSGSHLSGTFGPVNPSLNSTYDFMSTLFLEISSVFPDFYLHLGGDEVDFTCWKSNPNIQAFMKKKGF-TDFKQLESFYIQTLLDIVSDYDKGYVVWQEVFDNKVKVRPDTIIQVWREEMPVEYMLEMQDITRAGFRALLSAPWYLNRVKYGPDWKDMYKVEPLAFHGTPEQKALVIGGEACMWGEYVDSTNLVPRLWPRAGAVAERLWSSNLTTNIDFAFKRLSHFRCELVRRGIQAQPISVGCCEQEFEQT*A--T--SA--E----HPG-------G------C----CP---------L-SQ-LR--*A--------P---RR-V--LALR-E----Q-VP--G-Q---G-*SFT---------A-SRPGES---T---P----CP---C--APVT--TEKEAGA----GT--GV--Q---*R-----------------------S-MW-HF-------L--'
Refining With Semi-global Alignment
The alignment is very good except for the terminal segments. For instance, notice the sparse matched pairs in the first positions. This occurs because a global alignment attempts to force the matching all the way to the ends and there is point where the penalty for opening new gaps is comparable to the score of matching residues. In some cases it is desirable to remove the gap penalty added at the ends of a global alignment; this allows you to better match this pair of sequences. This technique is commonly known as 'semi-global' alignment or 'glocal' alignment.
[score, globalAlignment] = nwalign(humanProtein,mouseProtein,'glocal',true)score = 1.0413e+03
globalAlignment = 3x825 char array
'SCRRPAQSAARSRSLRSRPEVKGQGVGPPGVAGAEPPLVT*FADKSRGRRSPDQGLTWPAPSERGDQR-AMTSSRLWFSLLLAAAFAGRATALWPWPQNFQTSDQRYVLYPNNFQFQYDVSSAAQPGCSVLDEAFQRYRDLLFGSGSWPRPYLTGKRHTLEKNVLVVSVVTPGCNQLPTLESVENYTLTINDDQCLLLSETVWGALRGLETFSQLVWKSAEGTFFINKTEIEDFPRFPHRGLLLDTSRHYLPLSSILDTLDVMAYNKLNVFHWHLVDDPSFPYESFTFPELMRKGSYNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGIPGLLTPCYSGSEPSGTFGPVNPSLNNTYEFMSTFFLEVSSVFPDFYLHLGGDEVDFTCWKSNPEIQDFMRKKGFGEDFKQLESFYIQTLLDIVSSYGKGYVVWQEVFDNKVKIQPDTIIQVWREDIPVNYMKELELVTKAGFRALLSAPWYLNRISYGPDWKDFYIVEPLAFEGTPEQKALVIGGEACMWGEYVDNTNLVPRLWPRAGAVAERLWSNKLTSDLTFAYERLSHFRCELLRRGVQAQPLNVGFCEQEFEQT*APGTEEGAGCR*MV-VEPGFHCILA-R----GR--SPLPSCP-LPA-CPCA-WRERGRCWRSHSIK-SNVAFFYNKHGLPVFKKKSVNGVRVRAQPGWSQCLPLRSFKLRAGNETYSLCAVLPCL*AMSLPSHS*PYSRHLP*SSACSLHFCIISPRRWYMEKDVGAWRCSGQWGGLQTQPGHRRASPPCILIHLPPLELFSFGFLAASILYNHYLNIIKHILFS'
' : || | ||:: ||| |||||||:| ||||||||| :|| :||:||||||||:| |||||| || ||||||:|||:||||||||||| :::|::|| ||:||||||| ||::|:|||||||||||||||||| |||||||||||||||||||||||||||||||:|:|||||||||:|||||||||||||||||||||||||:|||||||||| |||||||||||| ||||:|||||||||||||||||||||||||||||||||||||||||| |||||||||||: ||||||||||||:||:||||:|||:|||||||||||||||||||||||||:|| ||:|||| ||||||||||||||||||:| |||||||||||||||::||||||||||::||:|| |:: :|:|||||||||||||||::|||||||:| ||||||:||||||||||||||||||||||:||||||||||||||||||||::||::: ||::|||||||||:|||:||||::|| |||||||||| ::|: :|| : :: : : :|| | |: | | | : |||| : : : ::: :: | '
'------------------------------------------------------------AAGRGAGRWAMAGCRLWVSLLLAAALACLATALWPWPQYIQTYHRRYTLYPNNFQFRYHVSSAAQAGCVVLDEAFRRYRNLLFGSGSWPRPSFSNKQQTLGKNILVVSVVTAECNEFPNLESVENYTLTINDDQCLLASETVWGALRGLETFSQLVWKSAEGTFFINKTKIKDFPRFPHRGVLLDTSRHYLPLSSILDTLDVMAYNKFNVFHWHLVDDSSFPYESFTFPELTRKGSFNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGAPGLLTPCYSGSHLSGTFGPVNPSLNSTYDFMSTLFLEISSVFPDFYLHLGGDEVDFTCWKSNPNIQAFMKKKGF-TDFKQLESFYIQTLLDIVSDYDKGYVVWQEVFDNKVKVRPDTIIQVWREEMPVEYMLEMQDITRAGFRALLSAPWYLNRVKYGPDWKDMYKVEPLAFHGTPEQKALVIGGEACMWGEYVDSTNLVPRLWPRAGAVAERLWSSNLTTNIDFAFKRLSHFRCELVRRGIQAQPISVGCCEQEFEQT*ATSAEHPGGCCPLSQLR*APRRVLALREQVPGQG*SFTASRPGESTPCPCAPVTTEKEAGAGTGVQ*RSMWHFL-------------------------------------------------------------------------------------------------------------------------------------------------------'
Refining the Alignment by Extracting the Protein Sequence
Another way to refine your alignment is by using only the protein sequences. Notice that the aligned region is delimited by start ( M-methionine ) and stop ( * ) amino acids in the sequences. If the sequence is shortened so that only the translated regions are considered, then it seems likely that you will get a better alignment. Use the find command to look for the index of the start amino acid in each sequence:
humanStart = find(humanProtein == 'M',1)humanStart = 70
mouseStart = find(mouseProtein == 'M',1)mouseStart = 11
Similarly, use the find command to look for the index of the first stop occurring after the start of the translation. Special care needs to be taken because there is also a stop at the very beginning of the humanProtein sequence.
humanStop = find(humanProtein(humanStart:end)=='*',1) + humanStart - 1humanStop = 599
mouseStop = find(mouseProtein(mouseStart:end)=='*',1) + mouseStart - 1mouseStop = 539
Use these indices to truncate the sequences.
humanSeq = humanProtein(humanStart:humanStop); humanSeqFormatted = seqdisp(humanSeq)
humanSeqFormatted = 9x70 char array
' 1 MTSSRLWFSL LLAAAFAGRA TALWPWPQNF QTSDQRYVLY PNNFQFQYDV SSAAQPGCSV'
' 61 LDEAFQRYRD LLFGSGSWPR PYLTGKRHTL EKNVLVVSVV TPGCNQLPTL ESVENYTLTI'
'121 NDDQCLLLSE TVWGALRGLE TFSQLVWKSA EGTFFINKTE IEDFPRFPHR GLLLDTSRHY'
'181 LPLSSILDTL DVMAYNKLNV FHWHLVDDPS FPYESFTFPE LMRKGSYNPV THIYTAQDVK'
'241 EVIEYARLRG IRVLAEFDTP GHTLSWGPGI PGLLTPCYSG SEPSGTFGPV NPSLNNTYEF'
'301 MSTFFLEVSS VFPDFYLHLG GDEVDFTCWK SNPEIQDFMR KKGFGEDFKQ LESFYIQTLL'
'361 DIVSSYGKGY VVWQEVFDNK VKIQPDTIIQ VWREDIPVNY MKELELVTKA GFRALLSAPW'
'421 YLNRISYGPD WKDFYIVEPL AFEGTPEQKA LVIGGEACMW GEYVDNTNLV PRLWPRAGAV'
'481 AERLWSNKLT SDLTFAYERL SHFRCELLRR GVQAQPLNVG FCEQEFEQT* '
mouseSeq = mouseProtein(mouseStart:mouseStop); mouseSeqFormatted = seqdisp(mouseSeq)
mouseSeqFormatted = 9x70 char array
' 1 MAGCRLWVSL LLAAALACLA TALWPWPQYI QTYHRRYTLY PNNFQFRYHV SSAAQAGCVV'
' 61 LDEAFRRYRN LLFGSGSWPR PSFSNKQQTL GKNILVVSVV TAECNEFPNL ESVENYTLTI'
'121 NDDQCLLASE TVWGALRGLE TFSQLVWKSA EGTFFINKTK IKDFPRFPHR GVLLDTSRHY'
'181 LPLSSILDTL DVMAYNKFNV FHWHLVDDSS FPYESFTFPE LTRKGSFNPV THIYTAQDVK'
'241 EVIEYARLRG IRVLAEFDTP GHTLSWGPGA PGLLTPCYSG SHLSGTFGPV NPSLNSTYDF'
'301 MSTLFLEISS VFPDFYLHLG GDEVDFTCWK SNPNIQAFMK KKGFTDFKQL ESFYIQTLLD'
'361 IVSDYDKGYV VWQEVFDNKV KVRPDTIIQV WREEMPVEYM LEMQDITRAG FRALLSAPWY'
'421 LNRVKYGPDW KDMYKVEPLA FHGTPEQKAL VIGGEACMWG EYVDSTNLVP RLWPRAGAVA'
'481 ERLWSSNLTT NIDFAFKRLS HFRCELVRRG IQAQPISVGC CEQEFEQT* '
Align these two sequences.
[score, alignment] = nwalign(humanSeq,mouseSeq)
score = 1.0423e+03
alignment = 3x530 char array
'MTSSRLWFSLLLAAAFAGRATALWPWPQNFQTSDQRYVLYPNNFQFQYDVSSAAQPGCSVLDEAFQRYRDLLFGSGSWPRPYLTGKRHTLEKNVLVVSVVTPGCNQLPTLESVENYTLTINDDQCLLLSETVWGALRGLETFSQLVWKSAEGTFFINKTEIEDFPRFPHRGLLLDTSRHYLPLSSILDTLDVMAYNKLNVFHWHLVDDPSFPYESFTFPELMRKGSYNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGIPGLLTPCYSGSEPSGTFGPVNPSLNNTYEFMSTFFLEVSSVFPDFYLHLGGDEVDFTCWKSNPEIQDFMRKKGFGEDFKQLESFYIQTLLDIVSSYGKGYVVWQEVFDNKVKIQPDTIIQVWREDIPVNYMKELELVTKAGFRALLSAPWYLNRISYGPDWKDFYIVEPLAFEGTPEQKALVIGGEACMWGEYVDNTNLVPRLWPRAGAVAERLWSNKLTSDLTFAYERLSHFRCELLRRGVQAQPLNVGFCEQEFEQT*'
'|:: ||| |||||||:| ||||||||| :|| :||:||||||||:| |||||| || ||||||:|||:||||||||||| :::|::|| ||:||||||| ||::|:|||||||||||||||||| |||||||||||||||||||||||||||||||:|:|||||||||:|||||||||||||||||||||||||:|||||||||| |||||||||||| ||||:|||||||||||||||||||||||||||||||||||||||||| |||||||||||: ||||||||||||:||:||||:|||:|||||||||||||||||||||||||:|| ||:|||| ||||||||||||||||||:| |||||||||||||||::||||||||||::||:|| |:: :|:|||||||||||||||::|||||||:| ||||||:||||||||||||||||||||||:||||||||||||||||||||::||::: ||::|||||||||:|||:||||::|| |||||||||'
'MAGCRLWVSLLLAAALACLATALWPWPQYIQTYHRRYTLYPNNFQFRYHVSSAAQAGCVVLDEAFRRYRNLLFGSGSWPRPSFSNKQQTLGKNILVVSVVTAECNEFPNLESVENYTLTINDDQCLLASETVWGALRGLETFSQLVWKSAEGTFFINKTKIKDFPRFPHRGVLLDTSRHYLPLSSILDTLDVMAYNKFNVFHWHLVDDSSFPYESFTFPELTRKGSFNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGAPGLLTPCYSGSHLSGTFGPVNPSLNSTYDFMSTLFLEISSVFPDFYLHLGGDEVDFTCWKSNPNIQAFMKKKGF-TDFKQLESFYIQTLLDIVSDYDKGYVVWQEVFDNKVKVRPDTIIQVWREEMPVEYMLEMQDITRAGFRALLSAPWYLNRVKYGPDWKDMYKVEPLAFHGTPEQKALVIGGEACMWGEYVDSTNLVPRLWPRAGAVAERLWSSNLTTNIDFAFKRLSHFRCELVRRGIQAQPISVGCCEQEFEQT*'
Open reading frame information is also available from the output of the seqshoworfs command, but the indices are based on the nucleotide sequences. Use these indices to trim the original nucleotide sequences and then translate them to amino acids.
humanPORF = nt2aa(humanHEXA.Sequence(humanORFs(1).Start(1):humanORFs(1).Stop(1))); mousePORF = nt2aa(mouseHEXA.Sequence(mouseORFs(1).Start(1):mouseORFs(1).Stop(1))); [score, ORFAlignment] = nwalign(humanPORF,mousePORF)
score = 1042
ORFAlignment = 3x529 char array
'MTSSRLWFSLLLAAAFAGRATALWPWPQNFQTSDQRYVLYPNNFQFQYDVSSAAQPGCSVLDEAFQRYRDLLFGSGSWPRPYLTGKRHTLEKNVLVVSVVTPGCNQLPTLESVENYTLTINDDQCLLLSETVWGALRGLETFSQLVWKSAEGTFFINKTEIEDFPRFPHRGLLLDTSRHYLPLSSILDTLDVMAYNKLNVFHWHLVDDPSFPYESFTFPELMRKGSYNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGIPGLLTPCYSGSEPSGTFGPVNPSLNNTYEFMSTFFLEVSSVFPDFYLHLGGDEVDFTCWKSNPEIQDFMRKKGFGEDFKQLESFYIQTLLDIVSSYGKGYVVWQEVFDNKVKIQPDTIIQVWREDIPVNYMKELELVTKAGFRALLSAPWYLNRISYGPDWKDFYIVEPLAFEGTPEQKALVIGGEACMWGEYVDNTNLVPRLWPRAGAVAERLWSNKLTSDLTFAYERLSHFRCELLRRGVQAQPLNVGFCEQEFEQT'
'|:: ||| |||||||:| ||||||||| :|| :||:||||||||:| |||||| || ||||||:|||:||||||||||| :::|::|| ||:||||||| ||::|:|||||||||||||||||| |||||||||||||||||||||||||||||||:|:|||||||||:|||||||||||||||||||||||||:|||||||||| |||||||||||| ||||:|||||||||||||||||||||||||||||||||||||||||| |||||||||||: ||||||||||||:||:||||:|||:|||||||||||||||||||||||||:|| ||:|||| ||||||||||||||||||:| |||||||||||||||::||||||||||::||:|| |:: :|:|||||||||||||||::|||||||:| ||||||:||||||||||||||||||||||:||||||||||||||||||||::||::: ||::|||||||||:|||:||||::|| ||||||||'
'MAGCRLWVSLLLAAALACLATALWPWPQYIQTYHRRYTLYPNNFQFRYHVSSAAQAGCVVLDEAFRRYRNLLFGSGSWPRPSFSNKQQTLGKNILVVSVVTAECNEFPNLESVENYTLTINDDQCLLASETVWGALRGLETFSQLVWKSAEGTFFINKTKIKDFPRFPHRGVLLDTSRHYLPLSSILDTLDVMAYNKFNVFHWHLVDDSSFPYESFTFPELTRKGSFNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGAPGLLTPCYSGSHLSGTFGPVNPSLNSTYDFMSTLFLEISSVFPDFYLHLGGDEVDFTCWKSNPNIQAFMKKKGF-TDFKQLESFYIQTLLDIVSDYDKGYVVWQEVFDNKVKVRPDTIIQVWREEMPVEYMLEMQDITRAGFRALLSAPWYLNRVKYGPDWKDMYKVEPLAFHGTPEQKALVIGGEACMWGEYVDSTNLVPRLWPRAGAVAERLWSSNLTTNIDFAFKRLSHFRCELVRRGIQAQPISVGCCEQEFEQT'
Alternatively, you can use the coding region information (CDS) from the GenBank data structure to find the coding region of the genes.
idx = humanHEXA.CDS.indices; humanCodingRegion = humanHEXA.Sequence(idx(1):idx(2)); idx = mouseHEXA.CDS.indices; mouseCodingRegion = mouseHEXA.Sequence(idx(1):idx(2));
You can also get the translation of the coding regions from this structure.
humanTranslatedRegion = humanHEXA.CDS.translation; mouseTranslatedRegion = mouseHEXA.CDS.translation;
Local Alignment
Instead of truncating the sequences to look for better alignment, an alternative approach is to use a local alignment. The function swalign performs local alignment using the Smith-Waterman algorithm. This shows a very good alignment for the whole coding region and reasonable similarity for a few residues beyond at both the ends of the gene.
[score, localAlignment] = swalign(humanProtein,mouseProtein)
score = 1057
localAlignment = 3x547 char array
'RGDQR-AMTSSRLWFSLLLAAAFAGRATALWPWPQNFQTSDQRYVLYPNNFQFQYDVSSAAQPGCSVLDEAFQRYRDLLFGSGSWPRPYLTGKRHTLEKNVLVVSVVTPGCNQLPTLESVENYTLTINDDQCLLLSETVWGALRGLETFSQLVWKSAEGTFFINKTEIEDFPRFPHRGLLLDTSRHYLPLSSILDTLDVMAYNKLNVFHWHLVDDPSFPYESFTFPELMRKGSYNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGIPGLLTPCYSGSEPSGTFGPVNPSLNNTYEFMSTFFLEVSSVFPDFYLHLGGDEVDFTCWKSNPEIQDFMRKKGFGEDFKQLESFYIQTLLDIVSSYGKGYVVWQEVFDNKVKIQPDTIIQVWREDIPVNYMKELELVTKAGFRALLSAPWYLNRISYGPDWKDFYIVEPLAFEGTPEQKALVIGGEACMWGEYVDNTNLVPRLWPRAGAVAERLWSNKLTSDLTFAYERLSHFRCELLRRGVQAQPLNVGFCEQEFEQT*APGTEEGAGC'
'|| | ||:: ||| |||||||:| ||||||||| :|| :||:||||||||:| |||||| || ||||||:|||:||||||||||| :::|::|| ||:||||||| ||::|:|||||||||||||||||| |||||||||||||||||||||||||||||||:|:|||||||||:|||||||||||||||||||||||||:|||||||||| |||||||||||| ||||:|||||||||||||||||||||||||||||||||||||||||| |||||||||||: ||||||||||||:||:||||:|||:|||||||||||||||||||||||||:|| ||:|||| ||||||||||||||||||:| |||||||||||||||::||||||||||::||:|| |:: :|:|||||||||||||||::|||||||:| ||||||:||||||||||||||||||||||:||||||||||||||||||||::||::: ||::|||||||||:|||:||||::|| |||||||||| ::|: :||'
'RGAGRWAMAGCRLWVSLLLAAALACLATALWPWPQYIQTYHRRYTLYPNNFQFRYHVSSAAQAGCVVLDEAFRRYRNLLFGSGSWPRPSFSNKQQTLGKNILVVSVVTAECNEFPNLESVENYTLTINDDQCLLASETVWGALRGLETFSQLVWKSAEGTFFINKTKIKDFPRFPHRGVLLDTSRHYLPLSSILDTLDVMAYNKFNVFHWHLVDDSSFPYESFTFPELTRKGSFNPVTHIYTAQDVKEVIEYARLRGIRVLAEFDTPGHTLSWGPGAPGLLTPCYSGSHLSGTFGPVNPSLNSTYDFMSTLFLEISSVFPDFYLHLGGDEVDFTCWKSNPNIQAFMKKKGF-TDFKQLESFYIQTLLDIVSDYDKGYVVWQEVFDNKVKVRPDTIIQVWREEMPVEYMLEMQDITRAGFRALLSAPWYLNRVKYGPDWKDMYKVEPLAFHGTPEQKALVIGGEACMWGEYVDSTNLVPRLWPRAGAVAERLWSSNLTTNIDFAFKRLSHFRCELVRRGIQAQPISVGCCEQEFEQT*ATSAEHPGGC'
Alignment of Complementary DNA Sequences
All the sequence alignment functions provided in MATLAB can be customized. For example, by modifying the rows and columns of a scoring matrix you can align sequences by complement and not by identity. In this case you can reorder the NUC44 scoring matrix; a positive score is given for complements while a negative score is given otherwise. The first 30 nucleotides from the mouse HEXA gene will be aligned to its complement.
[M, info] = nuc44; map = nt2int(seqcomplement(info.Order))
map = 1x15 uint8 row vector
4 3 2 1 6 5 8 7 9 10 14 13 12 11 15
Mc = M(:,map)
Mc = 15×15
-4 -4 -4 5 -4 1 1 -4 -4 1 -1 -1 -1 -4 -2
-4 -4 5 -4 1 -4 1 -4 1 -4 -1 -1 -4 -1 -2
-4 5 -4 -4 -4 1 -4 1 1 -4 -1 -4 -1 -1 -2
5 -4 -4 -4 1 -4 -4 1 -4 1 -4 -1 -1 -1 -2
-4 1 -4 1 -4 -1 -2 -2 -2 -2 -1 -3 -1 -3 -1
1 -4 1 -4 -1 -4 -2 -2 -2 -2 -3 -1 -3 -1 -1
1 1 -4 -4 -2 -2 -4 -1 -2 -2 -3 -3 -1 -1 -1
-4 -4 1 1 -2 -2 -1 -4 -2 -2 -1 -1 -3 -3 -1
-4 1 1 -4 -2 -2 -2 -2 -1 -4 -1 -3 -3 -1 -1
1 -4 -4 1 -2 -2 -2 -2 -4 -1 -3 -1 -1 -3 -1
⋮
[score, compAlignment] = nwalign(mouseHEXA.Sequence(1:30), ... seqcomplement(mouseHEXA.Sequence(1:30)), 'SCORINGMATRIX', ... Mc, 'ALPHABET', 'NT')
score = 150
compAlignment = 3x30 char array
'GCTGCTGGAAGGGGAGCTGGCCGGTGGGCC'
'::::::::::::::::::::::::::::::'
'CGACGACCTTCCCCTCGACCGGCCACCCGG'
close all;You can also select a web site from the following list:
Americas
- América Latina (Español)
- Canada (English)
- United States (English)
Europe
- Belgium (English)
- Denmark (English)
- Deutschland (Deutsch)
- España (Español)
- Finland (English)
- France (Français)
- Ireland (English)
- Italia (Italiano)
- Luxembourg (English)
- Netherlands (English)
- Norway (English)
- Österreich (Deutsch)
- Portugal (English)
- Sweden (English)
- Switzerland
- United Kingdom (English)